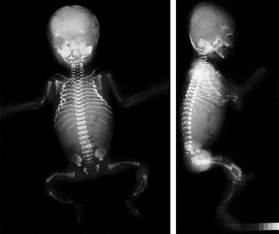
 X-Ray of a child with type III OI
X-Ray of a child with type III OI
By Cera Cruise
Imagine having bones so fragile that even a small cough could cause you to break a rib. That's what sufferers of type III osteogenesis imperfect (OI) have to deal with. Over their life time, a person with OI could have any were from a couple of fractures to several hundred.
So What Causes Osteogenesis Imperfecta?
Types I-IV are dominant forms of Osteogenesis Imperfecta that are caused by a mutation of the type 1 collagen (COL1A1 or COL1A2) genes that affects the body’s production of the collagen found in bones and other tissue. Type V's causes are unknown, but it too is a dominant form of OI. 85%-90% of cases of OI are caused by a dominant mutation. In order to have a dominant form of OI only one parent has to carry the gene mutation, or if neither parent is a carrier then the OI is caused by a spontaneous mutation. A person with a dominant form of OI has a 50% chance of passing it on to their children.
There are two, possibly three types of recessive OI, type VII and type VIII are definetly caused by a recessive mutation, while it is unknown whether type VI is caused by a dominant or recessive mutation, though it is presumed to be recessive. Types VII and VIII are caused by two separate mutations of either the cartilage-associated protein gene (CRTAP) or the prolyl 3-hydroxylase 1 gene (LEPRE1). In order to inherit a recessive form of OI both parents must be carriers and the child has to inherit a copy of the mutation from each parent. Only 10%-15% of cases of OI are recessive. Siblings of someone with a recessive form of OI have a 50% chance of being a carrier of that type of OI. 100% of children with a parent with a recessive form of OI will be carriers.
Imagine having bones so fragile that even a small cough could cause you to break a rib. That's what sufferers of type III osteogenesis imperfect (OI) have to deal with. Over their life time, a person with OI could have any were from a couple of fractures to several hundred.
So What Causes Osteogenesis Imperfecta?
Types I-IV are dominant forms of Osteogenesis Imperfecta that are caused by a mutation of the type 1 collagen (COL1A1 or COL1A2) genes that affects the body’s production of the collagen found in bones and other tissue. Type V's causes are unknown, but it too is a dominant form of OI. 85%-90% of cases of OI are caused by a dominant mutation. In order to have a dominant form of OI only one parent has to carry the gene mutation, or if neither parent is a carrier then the OI is caused by a spontaneous mutation. A person with a dominant form of OI has a 50% chance of passing it on to their children.
There are two, possibly three types of recessive OI, type VII and type VIII are definetly caused by a recessive mutation, while it is unknown whether type VI is caused by a dominant or recessive mutation, though it is presumed to be recessive. Types VII and VIII are caused by two separate mutations of either the cartilage-associated protein gene (CRTAP) or the prolyl 3-hydroxylase 1 gene (LEPRE1). In order to inherit a recessive form of OI both parents must be carriers and the child has to inherit a copy of the mutation from each parent. Only 10%-15% of cases of OI are recessive. Siblings of someone with a recessive form of OI have a 50% chance of being a carrier of that type of OI. 100% of children with a parent with a recessive form of OI will be carriers.
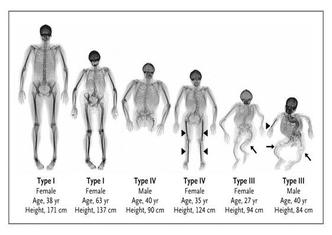
What are the different types of Osteogenesis
Imperfecta?
There are 8 different types of OI, with 5 of them definitely being caused by a dominant mutation. Here is a brief description of them;
Dominant Forms of Osteogenesis Imperfecta
Type I: Type I OI is the most common and mildest type of OI, most fractures occur before puberty. The sclerae (whites of eyes) usually have a blue, purple, or gray tint, and hearing loss is possible in the late 20's. Spinal curvature, dental issues and a triangular face are also symptoms. The structure of collagen is normal, but there is less of it in their bones. Type OI is highly manageable, and many people live their whole lives not even knowing they have it.
Type II: Type II OI is often fatal at birth or soon after, and the most severe type of OI. The collagen is improperly formed, and the lungs are underdeveloped due to a small stature with severe bone deformity and numerous fractures. The sclerae are either dark blue or gray.
Type III: Type III OI is the second most severe type of OI. By the time a baby with type III OI is born it will already have many broken bones, and possibly some healed fractures that occurred before birth. The collagen is improperly formed and at birth, infants generally have mildly shortened and bowed limbs, small chests tinted sclerae, and a soft skull cap. Because of their barrel shaped chests, people with type III OI often have repertory problems. Other symptoms include loose joints, muscle weakness, brittle teeth, hearing loss, and a triangular face.
Type IV: Type IV OI can range in severity from moderate to severe. In some cases it resembles Type I OI, while in others it resembles Type III OI, though there are key differences such as, people with Type IV OI tend to be born with blue sclerae, but by the time they reach puberty their sclerae are white. Bone deformity is normal, as is scoliosis, vertebrae compression, and ligament laxity. It is also common for the humerus and femur to be short, resulting in a short stature. Many cases of type IV OI are caused by a spontaneous mutation, and people with it have structurally defective type 1 collagen, which is present in reduced amounts.
There are 8 different types of OI, with 5 of them definitely being caused by a dominant mutation. Here is a brief description of them;
Dominant Forms of Osteogenesis Imperfecta
Type I: Type I OI is the most common and mildest type of OI, most fractures occur before puberty. The sclerae (whites of eyes) usually have a blue, purple, or gray tint, and hearing loss is possible in the late 20's. Spinal curvature, dental issues and a triangular face are also symptoms. The structure of collagen is normal, but there is less of it in their bones. Type OI is highly manageable, and many people live their whole lives not even knowing they have it.
Type II: Type II OI is often fatal at birth or soon after, and the most severe type of OI. The collagen is improperly formed, and the lungs are underdeveloped due to a small stature with severe bone deformity and numerous fractures. The sclerae are either dark blue or gray.
Type III: Type III OI is the second most severe type of OI. By the time a baby with type III OI is born it will already have many broken bones, and possibly some healed fractures that occurred before birth. The collagen is improperly formed and at birth, infants generally have mildly shortened and bowed limbs, small chests tinted sclerae, and a soft skull cap. Because of their barrel shaped chests, people with type III OI often have repertory problems. Other symptoms include loose joints, muscle weakness, brittle teeth, hearing loss, and a triangular face.
Type IV: Type IV OI can range in severity from moderate to severe. In some cases it resembles Type I OI, while in others it resembles Type III OI, though there are key differences such as, people with Type IV OI tend to be born with blue sclerae, but by the time they reach puberty their sclerae are white. Bone deformity is normal, as is scoliosis, vertebrae compression, and ligament laxity. It is also common for the humerus and femur to be short, resulting in a short stature. Many cases of type IV OI are caused by a spontaneous mutation, and people with it have structurally defective type 1 collagen, which is present in reduced amounts.
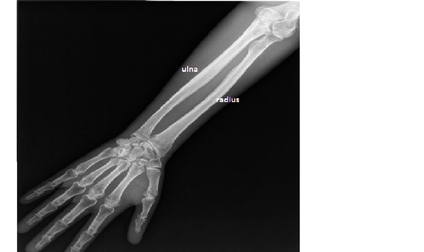
Types V and VI are both very similar to type IV OI, though the
cause of the mutation is unknown. It is only possible to differentiate them by
using x-rays and looking at the bones with a microscope.
Type V: When bones of someone with type V OI are studied the bones appear to have a mesh like appearance, while on x-rays dense bands are seen adjacent to the growth plates of the long bones. Hypertrophic calluses (bone growths) are commonly found at the sites of fractures or surgical procedures, as well as the calcification of the membrane between the radius and ulna leading to the restriction of forearm movement.
Type VI: It is unknown whether type VI OI is caused by a dominant or recessive mutation, though it is presumed to be a recessive mutation as type VI OI is extremely rare. People with this type of OI have slightly elevated alkaline phosphatase (enzyme linked to bone formation) activity level. When viewed under a microscope, bones have a fish scale appearance.
Recessive Forms of Osteogenesis Imperfecta
There are no mild cases of recessive OI, only moderate to fatal.
Type VII: Type VII can be fatal, as this type of OI is caused by mutation of the cartilage-associated protein gene (CRTAP). If someone has partial function of the CRTAP gene they will present with moderate symptoms, but if they don’t have the gene in all four cases it has proven to be fatal. Coxa Vara (deformity of the head of the femur) is common, as well as short stature. People with type VII OI have white sclerae.
Type VIII: Type VIII OI resembles type II or type III OI in appearance and symptoms except for the fact that infants have white sclerae. People with this form of OI suffer from severe growth deficiency. Type VIII OI is caused by a lack of P3H1 (propyl 3 hydroxylase 1) due to a mutation of the LEPRE1 gene.
Do people with OI have a normal lifespan?
Most people with OI lead long lives, though in severe cases their life span is shortened dramically. Respitory problems are the leading cause of death in people with OI, followed by accidental trauma.
Type V: When bones of someone with type V OI are studied the bones appear to have a mesh like appearance, while on x-rays dense bands are seen adjacent to the growth plates of the long bones. Hypertrophic calluses (bone growths) are commonly found at the sites of fractures or surgical procedures, as well as the calcification of the membrane between the radius and ulna leading to the restriction of forearm movement.
Type VI: It is unknown whether type VI OI is caused by a dominant or recessive mutation, though it is presumed to be a recessive mutation as type VI OI is extremely rare. People with this type of OI have slightly elevated alkaline phosphatase (enzyme linked to bone formation) activity level. When viewed under a microscope, bones have a fish scale appearance.
Recessive Forms of Osteogenesis Imperfecta
There are no mild cases of recessive OI, only moderate to fatal.
Type VII: Type VII can be fatal, as this type of OI is caused by mutation of the cartilage-associated protein gene (CRTAP). If someone has partial function of the CRTAP gene they will present with moderate symptoms, but if they don’t have the gene in all four cases it has proven to be fatal. Coxa Vara (deformity of the head of the femur) is common, as well as short stature. People with type VII OI have white sclerae.
Type VIII: Type VIII OI resembles type II or type III OI in appearance and symptoms except for the fact that infants have white sclerae. People with this form of OI suffer from severe growth deficiency. Type VIII OI is caused by a lack of P3H1 (propyl 3 hydroxylase 1) due to a mutation of the LEPRE1 gene.
Do people with OI have a normal lifespan?
Most people with OI lead long lives, though in severe cases their life span is shortened dramically. Respitory problems are the leading cause of death in people with OI, followed by accidental trauma.
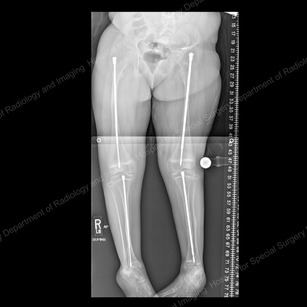 Rodding surgery can help prevent
bone deformity as shown is this X-ray
Rodding surgery can help prevent
bone deformity as shown is this X-ray
Is there a treatment for Osteogenesis Imperfecta?
Unfortunately there is no cure for OI, but there are many ways to strengthen bones and help manage the disease
Non-surgical treatments
Medical bisphosphonates have been proven to help reduce the amount of fractures and bone pain as well as slow down bone resorption. Walkers, braces, and wheelchair can assist in mobility, while physical therapy and light exercise such as swimming can help with muscle development.
Surgical treatments
There are many surgical options that can help with OI, such as surgery to manage; recurring fractures, bowing of the bones, scoliosis (sideways curvature of the spine), and a child's ability to sit or stand. Rodding (minimally-invasive procedure to insert a simple or telescopic metal rod the length of a long bone to stabilize it and prevent deformity) is another surgical procedure that can help dramatically.
Unfortunately there is no cure for OI, but there are many ways to strengthen bones and help manage the disease
Non-surgical treatments
Medical bisphosphonates have been proven to help reduce the amount of fractures and bone pain as well as slow down bone resorption. Walkers, braces, and wheelchair can assist in mobility, while physical therapy and light exercise such as swimming can help with muscle development.
Surgical treatments
There are many surgical options that can help with OI, such as surgery to manage; recurring fractures, bowing of the bones, scoliosis (sideways curvature of the spine), and a child's ability to sit or stand. Rodding (minimally-invasive procedure to insert a simple or telescopic metal rod the length of a long bone to stabilize it and prevent deformity) is another surgical procedure that can help dramatically.

Living with Osteogenesis Imperfecta
Everyday life
Depending on the severity, living with OI can be challenging. As a parent with a child with OI it can be extremely stressful, with severe cases even lifting your child’s ankle when changing their diaper can break their leg. All members of a family are affected by having someone in the family with OI. It can cause many issues with siblings who feel unloved due to lack of attention, as well as guilt if they accidently break their sibling’s bone, which is bound to happen at some point if their sibling’s OI is severe.
It can be hard to make strong friendships when you have OI, especially if any type of physical activity puts you at risk of injury. Because of their small stature, some people with OI are treated younger than they are which can be frustrating. Reoccurring hospitatil visits can be tough, especially for children. A broken bone could mean being immobile for the next month, or even having to have surgery.
Frequently child abuse allegations are brought upon parents with a child with OI if they don’t have a doctor’s note stating their child has OI. This can be traumatizing for both the child and parents.
However most people with OI lead productive lives, have successful careers, and raise their own families. OI is becoming more and more managable, and hopefully in the years to come a cure will be found.

If you are interested in having more of an insite into the lives of people with OI I highly suggest you read the book Handle with Care by Jodi Picoult. It is about a young girl named Willow who has type III OI, and describes some of the troubles that families face when a member has OI.
